Computational Protein Design to Address Challenges in Biotechnology
Over the last two years research in my group focused on developing methods to design and functionalize de novo proteins. Ultimately, we are aiming to be able to routinely and robustly design catalytic or small molecule binding proteins of arbitrary shapes.
DE NOVO PROTEIN DESIGN
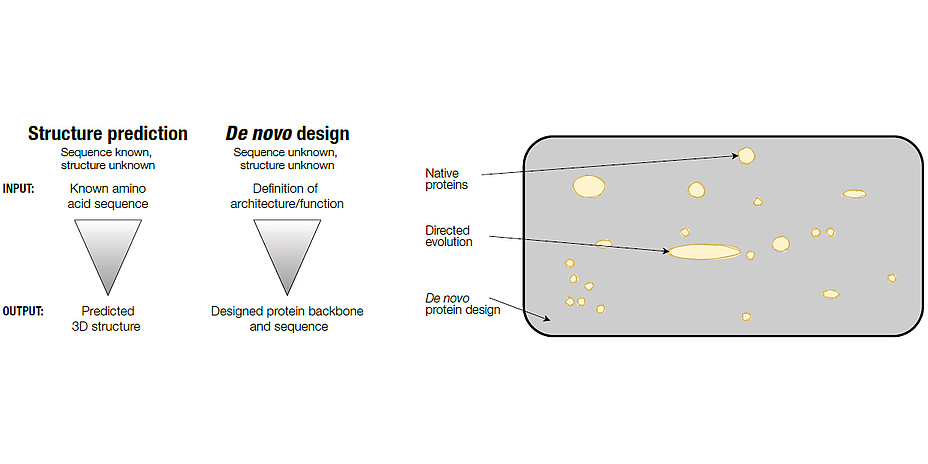
Since the beginning of protein science, it has been clear that proteins have tremendous potential to tackle and solve a variety of biomedical and biotechnological challenges. This is the major reason why they are used widely e.g. as drugs to treat diseases or to generate highly pure chemical compounds, while at the same time producing only minimal amounts of waste and exhibiting an excellent resource balance. In addition, researchers have long shown that the element that gives rise to a protein’s function is its three-dimensional structure – commonly referred to as the structure-function relationship – and that all the information needed to adopt this function/structure is stored and encoded in the amino acid sequence of the protein. It was proposed as early as 1963 by Christian Anfinsen that the structure a protein adopts is always the lowest energy state accessible to its amino acid sequence. Remarkably, with all the given degrees of freedom, even for a small protein, this process would take a prohibitively long time, if all the conformations it could adopt were to be explored (referred to as Levinthal’s paradox or the protein folding problem). This implies, however, that rather than being a set of random trials, the folding trajectory of a protein follows a path. So, if we can come up with ways to simulate and solve this problem efficiently, we should be able to make proteins from scratch according to our needs.
De novo protein design is the attempt to use our best understanding of protein biochemistry and biophysics – how proteins fold into their shapes by burial of hydrophobic amino acid residues, or what the typical inter- and intramolecular interactions of amino acids are and how they interact with their environment or targets/substrates – to identify a minimum energy amino acid sequence composition that allows the protein to fold exactly into a desired shape. This is essentially the protein structure prediction problem turned upside down, where a minimum energy structure for a given amino acid sequence is computed. Computationally, protein design represents two interconnected problems: a) How do we score conformations of an amino acid chain and b) How can we sample all its degrees of freedom efficiently? These problems are difficult to solve because sequence space for a typically sized protein (~200 amino acids) is vast (20200) and comprehensive sampling of it remains a challenge even with current computational power. Besides that, the free energy of such a large system is very difficult to compute with absolute accuracy.
Exploring Sequence Space
So why try to design a protein, if the odds of success are against the experimenter? With the advent of protein sequence databases and their ever-increasing growth, it has become evident that nature only sampled an infinitesimal small subset of all possible sequences available. Protein design on the contrary allows for the exploration of this “dark matter” of amino acid sequence space (Figure 1). However, the question remaining is: Is it possible to find something new in this pool of unexplored sequences? Given the sheer number of available and yet unexplored sequences, it is reasonable to argue that there are thousands of possibilities for designing novel proteins of high stability and arbitrary shape. All of these bare the potential to go beyond classical biochemical approaches and could ultimately provide solutions to biomedical and biotechnological challenges much faster than nature can. Over the last couple of years, tremendous progress has been made in this direction with many novel protein structures designed from scratch. This can be attributed to advances in understanding the fundamental processes underlying protein folding and concomitant improvements in computational methods. In addition, breakthroughs in the field of synthetic DNA manufacturing and the increase in computational power were key aspects for these successes.

Figure 2: Computationally designed helix-helix interface. Through iterative fine sampling of backbone geometries, the computations converged on “ideal” knobs-into-holes packing arrangements, without enforcing sequence motifs to achieve these types of interactions.

Figure 3: Side and top view of a computationally designed 20-helix bundle. This topology is completely unknown in nature.
Functionalizing Helical De Novo Proteins By Deviating From Ideal Geometries
Coiled-coils, a particular group of protein structures, have seen big advances in terms of design. These usually parallel and oligomeric protein assemblies present ideal targets for protein design studies, as they are very regular and follow a repeating sequence, which, in the canonical case, is seven residues long. We could show that it is possible to design genetically encoded, single-chain helix bundle structures with atomic level accuracy. To do so, a novel method that uses equations originally derived by Francis Crick in 1953 which accurately describe the geometries of -helical protein structures was established and used to sample the folding space of helical proteins computationally. The resulting designed proteins were highly idealistic in terms of geometry and showed very high thermodynamic stability (extrapolated ΔGfold > 60 kcal mol-1), with their experimentally determined structures close to identical to the design models and nearly perfect packing of amino acid side chains between the helices (referred to as “knobs-into-holes”, Figure 2). However, it is obvious that in nature, most protein functional sites sit at the end of structural elements or in unstructured regions and therefore are not placed at positions of ideal protein geometry. It has been shown that this can be a result of selective pressure, where the ancestral proteins had more regular structural elements, exhibited higher thermo-dynamic stabilities and less dynamics, in comparison to their contemporary versions. This is why it is still unclear whether idealized protein structures can be functionalized. In order to address this question, research in my group is currently focusing on designing large proteins with topologies not observed in nature. Key elements we hope to find with these studies are whether they exhibit similar rigidity and stability as observed for the small ideal proteins we designed previously (Figure 3). We are also investigating to what degree we can harness some of the very high thermo-dynamic stability of our parametrically designed helical bundles to introduce deviations from ideal geometry for the gain of catalytic function. To test different levels of deviation in helical backbones and to check if this is concomitant with a reduction of thermo-dynamic stability, functional sites of various sizes have been chosen. In particular my lab is working on metal complexation and cofactor binding (Figure 4). The ability to sample hundreds of thousands of potential protein backbones which can be used as starting points to introduce catalytic or ligand binding sites into de novo designed helical proteins is a big advantage over previous attempts in designing functional proteins. Initial results from this research show that there might be a tradeoff between high stability and degree of idealism as far as the protein backbone is concerned; however, many more designs have to be made and characterized before we can draw definite conclusions. In answering these questions though, we hope to pave the way for downstream applications of de novo protein design to tackle environmental, biomedical and biotechnological problems.

Figure 4: Computationally designed 4-helix bundle with a designed binding site for heme B. It can clearly be seen how much the helices had to be bent, in order to accommodate the heme.
This research area is anchored in the Field of Expertise “Human & Biotechnology”, one of five strategic foci of TU Graz.
You can find more research news on Planet research. Monthly updates from the world of science at Graz University of Technology are available via the research newsletter TU Graz research monthly.
Kontakt
Gustav OBERDORFER
Ass.Prof. Mag.rer.nat. Dr.rer.nat.
Institute of Biochemistry
Petersgasse 10-12
8010 Graz
Phone: +43 316 873 6462
gustav.oberdorfer@tugraz.at


