
Molekulardynamik-Simulationen der Leerstellendiffusion und Extraktion kontinuumsmechanischer Kennwerte
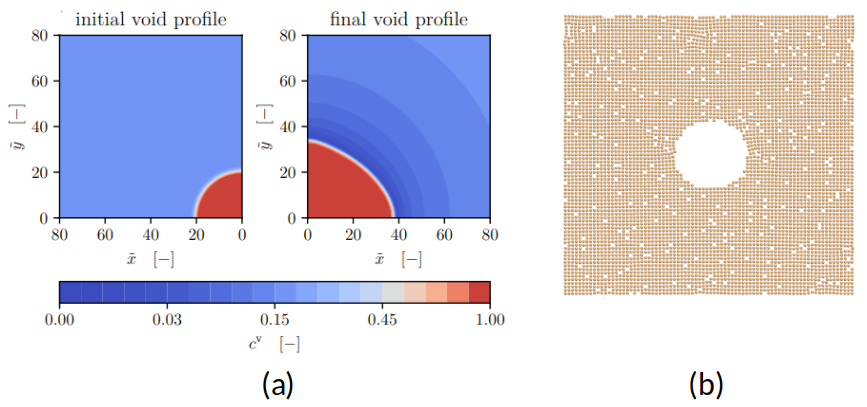
(b) Atomistische Darstellung der Anfangskonfiguration
Kontinuumsmodelle sagen voraus, dass Leerstellen in Kristallen entlang von Spannungsgradienten in Bereiche niedrigerer hydrostatischer Spannung driften. Dieses Verhalten prägt auf mesoskopischer Ebene die Phasenfeldbeschreibung von Porenbildung und -wachstum; ein bestehendes Phasenfeldmodell [1] zeigt dabei anisotropes Porenwachstum unter Zugbelastung. Ziel dieser Arbeit ist das Untersuchen dieses Phänomens auf mikroskopischen Skalen durch atomistische Simulationen und das Ableiten von Kenngrößen, mit denen das Phasenfeld-Modell bereichert werden soll. Atomistische Simulationen (Molekulardynamik, MD) verfolgen die Bewegung einzelner Atome anhand realistischer Potentiale. Ihr Vorteil ist die Einsicht in atomare Sprungereignisse zwischen Gitterplätzen, die zu Diffusion führen, in die zugehörigen Aktivierungsbarrieren und in die lokale Kopplungen zwischen Defekten und Spannung – ohne modellhafte Vorannahmen der Kontinuumsskala.
Im Zuge der Masterarbeit sollen MD-Simulationen der Leerstellendiffusion unter definierten Spannungszuständen mithilfe des Softwarepakets LAMMPS [2] untersucht und diese mit bekannten Ergebnissen aus Diffusions- und Phasenfeldmodellen abgeglichen werden. Die gewonnenen atomistischen Ergebnisse sollen zur Ableitung von Kontinuumsgrößen verwendet und zur Interpretation von Indikatoren verwendet werden, die als Eingangsparameter für mesoskopische Phasenfeldmodelle dienen (wie etwa effektive Diffusivität/Mobilität).
[1] K. A. Pendl and T. Hochrainer. Coupling stress fields and vacancy diffusion in phase-field models of voids as pure vacancy phase. Comput. Mater. Sci., 224 (2023)
[2] A. P. Thompson et al. LAMMPS - a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales. Comput. Phys. Commun., 271 (2022)
Aufgaben:
• Literaturrecherche Molekulardynamik und Leerstellenbewegung (unter mechanischer Belastung) sowie Leerstellenbewegung in kontinuumsmechanischen Diffusionsmodellen
• Einarbeitung in LAMMPS (Überblick und einfache Tests)
• Implementierung der Simulations- und Post-Processing-Pipelines (Setups mit definierter Spannung/Deformation; Defekte einbringen; Parameterstudie Kontinuumspunkt; Auswertung)
• Validierung der Algorithmen, Analyse und Dokumentation der Ergebnisse
Was Sie mitbringen sollen:
• Interesse an Festkörpermechanik und gekoppelten Problemstellungen
• Programmiererfahrung (mit einer beliebigen Programmiersprache) vorteilhaft
• Bereitschaft sich in vorhandene Softwarepakete (LAMMPS) einzuarbeiten
Was wir Ihnen bieten:
• Intensive Betreuung am Institut
• Einarbeiten in numerische Algorithmen und Anwenden von theoretischem Wissen
Beginn: ab sofort möglich
Kontakt: Dipl.-Ing. Dr.techn. Kevin Pendl BSc kevin.pendl@tugraz.at
Betreuer: Assoc.Prof. Dipl.-Ing. Dr.techn. Manfred Ulz & Dipl.-Ing. Dr.techn. Kevin Pendl BSc