Zincdibromide
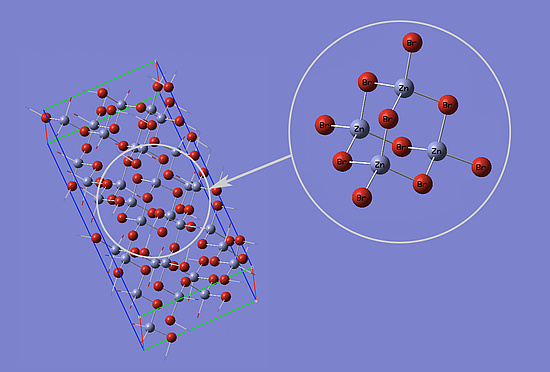
Fig. 1 shows a unit cell (left) and a subelement (right) of a ZnBr2 crystal. The unit cell contains 96 atoms and 3600 electrons. This large amount of electrons makes simulations difficult. Because of the limited operative memory simple basis sets are required.
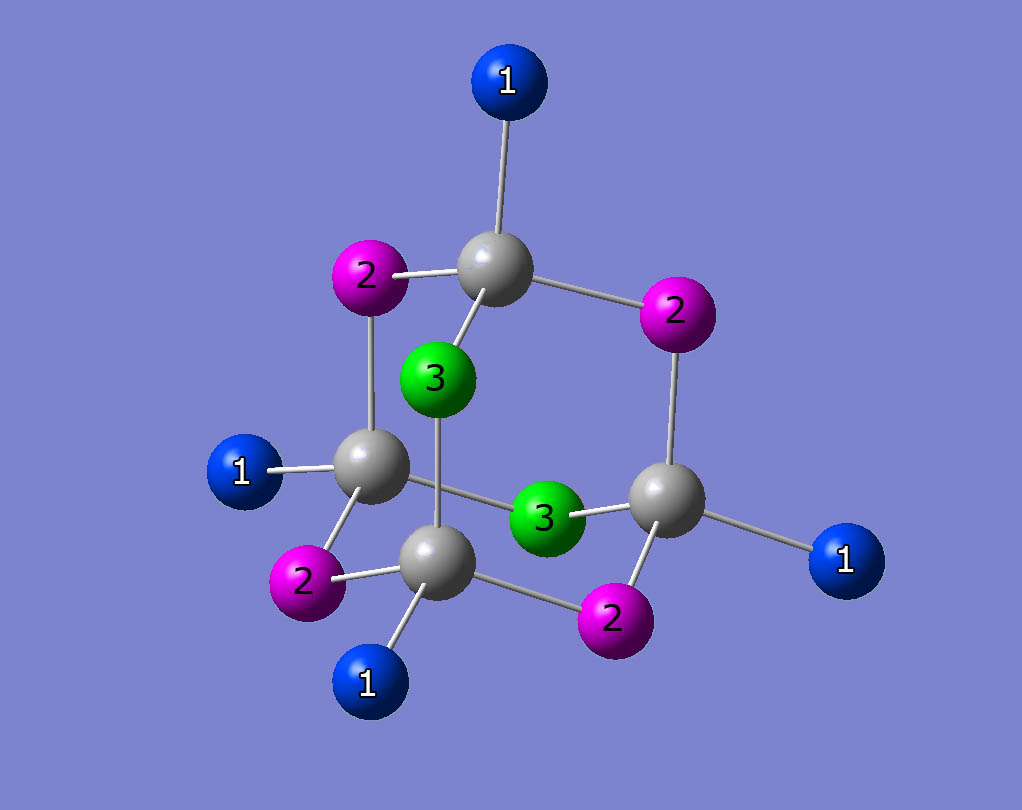
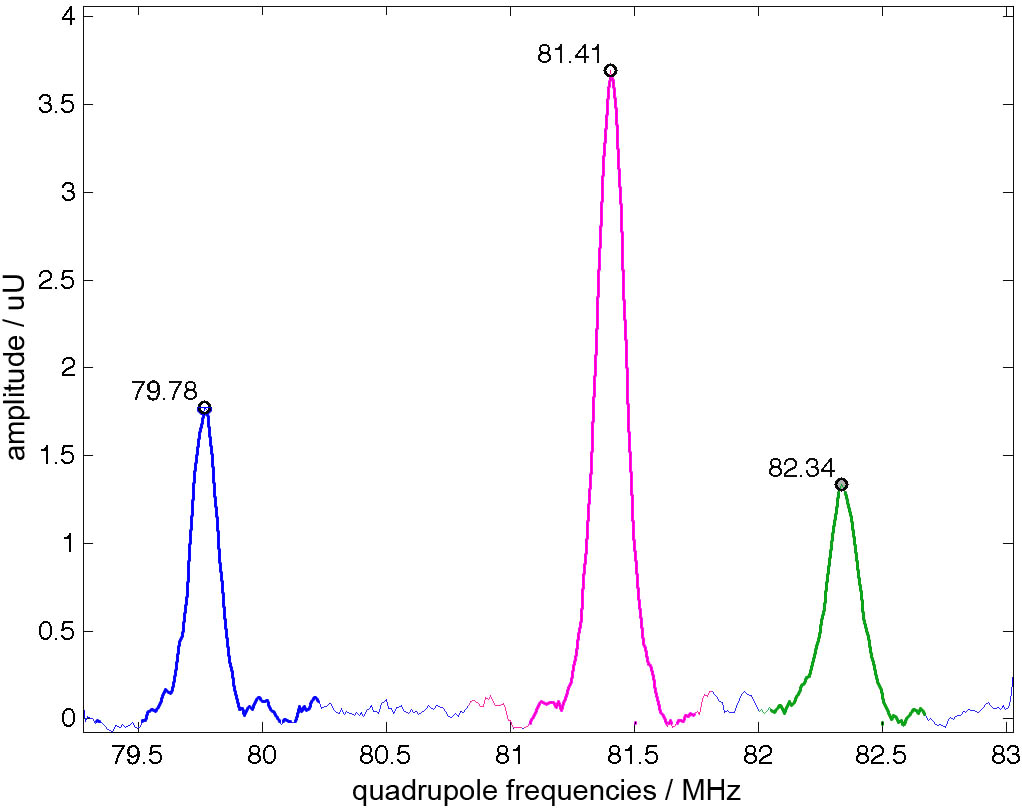
The Br atoms of ZnBr2 are located at three different crystallographic sites. This results in three different quadrupolar frequencies per isotope (79Br, 81Br). Figure 2 shows a ZnBr2 subelement. The three different crystallographic sites are coloured in green, blue and magenta. Figure 3 shows a nuclear quadrupole resonance spectrum. The three peaks correspond to the three different crystallographic sites shown in fig. 2. All results refer to 81Br.
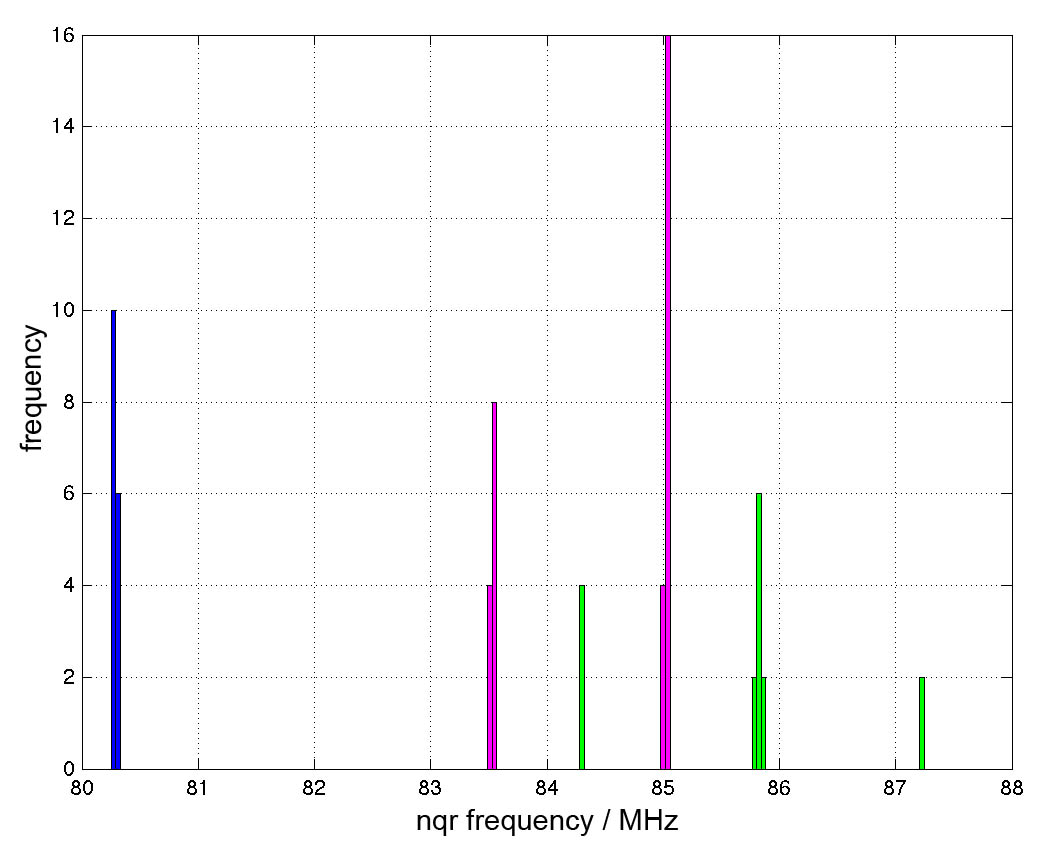
Due to the different crystallographic sites of Br atoms accurate results are just expected if the whole unit cell is simulated. Because of the large unit cell simple basis sets were chosen. Figure 4 shows a histogram of simulated frequencies. The simulated frequencies of the blue coloured crystallographic site have a small standard derivation and fit quiet good to the measured data. The magenta and the green group have a larger standard derivation and show a bigger difference to the measured data. Nevertheless are the simulated results near to the measured frequencies. Considering the limitations in choosing the basis set, the results are very good.